

CUT&RUN Assay Kit (with Drosophila Spike-In Control) 实验步骤
| ! | ! 表示实验步骤中需要 根据进行的 CUT&RUN 反应次数来 调整体积的重要步骤。 |
| !! | !! 表示需要在操作前稀释 缓冲液 的一个重要步骤。 |
| 安全停止 | 如果需要停止,这是实验步骤中的一个安全停止点。 |
一、Concanavalin A 珠子的激活
开始之前:
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
将 Concanavalin A Bead Activation Buffer 放在冰上。
- 确定要进行的 CUT&RUN 反应次数。我们强烈建议包含阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 和阴性对照 Rabbit (DA1E) Monoclonal Antibody IgG Isotype Control (CUT&RUN) #66362 的反应。
- 通过轻轻上下吸打小心地重悬 Concanavalin A 磁珠,确保不要将任何磁珠悬浮液溅出试管。
- 对于每 CUT&RUN 反应,将 10 µL 珠子悬液转移到一个新的 1.5 mL 微量离心管中。如果计划一次进行 14 次以上的 CUT&RUN 反应,请使用两个或更多个 1.5 mL 微量离心管。每 1.5 mL 微量离心管中应加入不超过 140 µL 的 Concanavalin A 珠子。
- 每 10 µL 珠子添加 100 µL Concanavalin A Bead Activation Buffer。上下吸打来轻轻混合珠子。
- 将试管放在磁力架上,直到溶液变澄清(30 秒至 2 分钟),然后去除并丢弃液体。
- 从磁力架上取下试管。重复步骤 4 和 5 一次,再次清洗珠子。
- 添加与最初添加的珠子悬液体积相等的 Concanavalin A Bead Activation Buffer(每份样品 10 µL),并通过上下吸打重悬。 注:活化的珠子可在冰上储存长达 8 小时。
注:避免将 Concanavalin A 磁珠悬液涡旋,因为反复涡旋可能会使 Concanavalin A 从珠子中移位。
注:为避免丢失珠子,在整个实验步骤中请勿使用真空来抽吸。
二、细胞和组织制备
对于大多数细胞类型,活细胞可用于 CUT&RUN 检测,以产生强大的组蛋白、转录因子和辅因子富集。就某些对 Concanavalin A 脆弱或敏感的细胞类型,略微固定细胞有助于保存细胞并保持它们完整。此外,如果使用新鲜细胞未观察到强大的信号,固定可能有助于促进低丰度和/或弱结合转录因子和辅因子的富集。请注意,细胞的过度固定会抑制 CUT&RUN 检测。
我们的 CUT&RUN 测定适用于广泛的细胞或组织输入。如实验步骤中定义,一次 CUT&RUN 反应可包含 5,000 至 250,000 个细胞或 1 至 5 mg 的组织。整个实验步骤中使用的缓冲液体积无需根据每反应的细胞或组织数量进行调整,只要该量在此范围内即可。当需要时,缓冲液体积确实需要根据正在执行的反应数量按比例增加。如果可能,我们建议每反应使用 100,000 个细胞或 1 mg 组织。如果细胞数量有限,我们建议每反应至少使用 5,000 到 10,000 个细胞进行组蛋白修饰检测,每反应至少使用 10,000 到 20,000 个细胞进行转录因子或辅因子检测。
A. 活细胞制备
开始之前:
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
加热 Protease Inhibitor Cocktail (200X) #7012 (PIC) 和 100X Spermidine #27287。确保两种物质完全融化。请注意,由于含有 DMSO,Protease Inhibitor Cocktail (200X) #7012 置于冰上时会重新冻结。
制备 1X 洗涤缓冲液(每种细胞系 2 mL,每个反应或每份输入样品额外制备 100 µL)。例如,要制备 2.5 mL 的 1X 洗涤缓冲液,加入 250 µL 10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 + 25 µL 100X Spermidine #27287 + 12.5 µL Protease Inhibitor Cocktail (200X) #7012 + 2,212.5 µL Nuclease-free Water #12931。让其平衡到室温,以最大程度减少细胞应激。
注:应在室温下连续进行活细胞(无固定)制备步骤,以尽量减少对细胞的压力。为了尽量减少 DNA 碎片,在重悬过程中避免剧烈涡旋和样品产生气泡。
- 在室温下收集新鲜的细胞培养物,以最大程度减少细胞应激。为每次反应收集 5,000 至 100,000 个细胞,并为输入样品额外收集 5,000 至 100,000 个细胞。确保包含阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 和阴性对照 Rabbit (DA1E) Monoclonal Antibody IgG Isotype Control (CUT&RUN) #66362 的反应。
- 将细胞悬液在室温下以 600 x g 的离心力离心 3 分钟,然后去除并丢弃液体。
- 在室温下,用 1 mL 1X 洗涤缓冲液(+ 亚精胺 + PIC)通过轻轻上下吸打来重悬细胞沉淀物。
- 在室温下以 600 x g 的离心力离心 3 分钟,然后去除并丢弃液体。
- 重复步骤 3 和 4 一次,再次洗涤细胞沉淀物。
- 对于每次反应或每份输入样品,添加 100 µL 1X 洗涤缓冲液(+ 亚精胺 + PIC),并轻轻上下吸打来重悬细胞沉淀物。
- 转移 100 µL 细胞到一个新试管中,置于 4°C 下储存,直至进行到第七部分。这是输入样品。
- 立即进入第三部分。
注:对于贴壁细胞,首先需要使用胰蛋白酶将细胞从培养皿中分离出来,并用至少 3 倍体积的组织培养基中和。我们不建议从培养皿中刮取细胞,因为这会压迫甚至裂解细胞。应使用血细胞计数器或其他细胞计数器对细胞进行计数,以确保用于实验的细胞数量正确。
注:如果处理的总细胞量少于 100,000 个,且离心后的细胞沉淀物肉眼不可见,则在洗涤步骤中易导致细胞丢失。因此,当处理低细胞量时,我们建议跳过下面的洗涤步骤 3 到 5。Concanavalin A 珠子与细胞的结合对结合反应中含有 40% 细胞培养基具有耐受性。因此,在步骤 2 中对细胞悬液进行初始离心后,可以去除并丢弃大部分上清液,每反应留下 ≤ 40 µL 细胞培养基。然后在步骤 6 中,向细胞悬液中加入足够的 1X 洗涤缓冲液(+ 亚精胺 + PIC),使每反应的总体积达到 100 µL。
注:在随后的实验步骤中,将在 55°C 下孵育输入样品,因此建议使用一根可封盖的 1.5 mL 试管,以减少孵育过程中的蒸发。
B. 固定细胞制备
注:固定细胞制备需要以下试剂,这些试剂不包含在此试剂盒中:37% 甲醛或 16% Formaldehyde, Methanol-Free #12606、Glycine Solution (10X) #7005 和 10% SDS Solution #20533。
开始之前:
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
加热 Protease Inhibitor Cocktail (200X) #7012 和 100X Spermidine #27287。确保两种物质完全融化。请注意,由于含有 DMSO,Protease Inhibitor Cocktail (200X) #7012 置于冰上时会重新冻结。
制备 1X 洗涤缓冲液(每种细胞系 2 mL,每次反应或每份输入样品额外制备 100 µL)。例如,要制备 2.5 mL 的 1X 洗涤缓冲液,加入 250 µL 10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 + 25 µL 100X Spermidine #27287 + 12.5 µL Protease Inhibitor Cocktail (200X) #7012 + 2,212.5 µL Nuclease-free Water #12931。让其平衡到室温,以最大程度减少细胞应激。
对于每 1 mL 待处理的细胞悬液,制备 2.7 µL 37% 甲醛或 6.25 µL 16% Formaldehyde, Methanol-Free #12606 并置于室温下保存。使用在制造商标示的有效期内的新鲜甲醛。
- 为每次反应收集 5,000 至 100,000 个细胞,并为输入样品额外收集 5,000 至 100,000 个细胞。确保包含阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 和阴性对照 Rabbit (DA1E) Monoclonal Antibody IgG Isotype Control (CUT&RUN) #66362 的反应。
- 每 1 mL 细胞悬液添加 2.7 µL 37% 甲醛或 6.25 µL 16% Formaldehyde, Methanol-Free #12606,以实现 0.1% 的最终浓度。旋转试管混匀并置于室温下孵育 2 分钟。
- 每 1 mL 固定细胞悬液添加 100 µL Glycine Solution (10X) #7005 以停止交联。旋转试管混匀并置于室温下孵育 5 分钟。
- 在 4°C 下将细胞悬液以 3,000 x g 的离心力离心 3 分钟,然后去除并丢弃液体。立即进入步骤 5。(安全停止)或者,固定细胞沉淀物可以在使用前于 -80°C 下储存长达 6 个月。
- 用 1 mL 1X 洗涤缓冲液(+ 亚精胺 + PIC)通过轻轻上下吸打来重悬细胞沉淀物。
- 在 4°C 下以 3,000 x g 的离心力离心 3 分钟,然后去除并丢弃液体。
- 重复步骤 5 和 6 一次,再次洗涤细胞沉淀物。
- 对于每次反应或每份输入样品,添加 100 µL 1X 洗涤缓冲液(+ 亚精胺 + PIC),并轻轻上下吸打来重悬细胞沉淀物。
- 转移 100 µL 细胞到一个新试管中,置于 4°C 下储存,直至进行到第七部分。这是输入样品。
- 立即进入第三部分。
注:对于贴壁细胞系,首先需要使用胰蛋白酶将细胞从培养皿中分离出来,并用至少 3 倍体积的培养基中和。我们不建议从培养皿中刮取细胞,因为这可能会对细胞造成应激,甚至导致细胞裂解。应使用血细胞计数器或其他细胞计数器对细胞进行计数,以确保用于实验的细胞数量正确。
注:如果处理的总细胞量少于 100,000 个,且离心后的细胞沉淀物肉眼不可见,则在洗涤步骤中易导致细胞丢失。在这种情况下,我们不建议冷冻细胞沉淀物。此外,当处理这些低细胞量时,我们建议跳过下面的洗涤步骤 5 到 7。Concanavalin A 珠子与细胞的结合对结合反应中含有 40% 细胞培养基具有耐受性。因此,在步骤 4 中对细胞悬液进行初始离心后,可以去除并丢弃大部分上清液,每反应留下 ≤ 40 µL 细胞培养基。然后在步骤 8 中,向细胞悬液中加入足够的 1X 洗涤缓冲液(+ 亚精胺 + PIC),使每反应的总体积达到 100 µL。
注:在随后的实验步骤中,将在 55°C 下孵育输入样品,因此建议使用一根可封盖的 1.5 mL 试管,以减少孵育过程中的蒸发。
C. 组织样品制备
对于大多数组织类型,1 mg 轻微固定的组织(0.1% 甲醛,2 分钟)可以产生强大的组蛋白、转录因子和辅因子富集。富集组蛋白修饰不需要甲醛固定。然而,许多转录因子和辅因子确实需要对组织进行轻微固定以获得最佳结果。一些低丰度和/或弱结合转录因子和辅因子可能需要中等固定(0.1% 甲醛,10 分钟)以获得最佳结果。此外,在使用具有挑战的组织类型(如纤维组织)时,中等固定可能会改善结果。请注意,过度固定会抑制 CUT&RUN 检测。固定的组织样品可以在使用前于 -80°C 下冷冻长达 6 个月。
注:固定组织制备需要以下试剂,这些试剂不包含在此试剂盒中:37% 甲醛或 16% Formaldehyde, Methanol-Free #12606、Phosphate Buffered Saline (PBS-1X) pH7.2 (Sterile) #9872、Glycine Solution (10X) #7005 和 10% SDS Solution #20533。
开始之前:
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
加热 Protease Inhibitor Cocktail (200X) #7012 和 100X Spermidine #27287。确保两种物质完全融化。请注意,由于含有 DMSO,Protease Inhibitor Cocktail (200X) #7012 置于冰上时会重新冻结。
- 制备 1X 洗涤缓冲液(每种组织类型 3 mL,每次反应或每份输入样品额外制备 100 µL)。例如,要制备 3.5 mL 的 1X 洗涤缓冲液,加入 350 µL 10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 + 35 µL 100X Spermidine #27287 + 17.5 µL Protease Inhibitor Cocktail (200X) #7012 + 3,097.5 µL Nuclease-free Water #12931。让其平衡到室温,以最大程度减少细胞应激。
如果需要组织固定,请准备以下缓冲液:
- 为每种组织类型制备 1 mL 固定缓冲液,方法如下:添加 2.7 µL 37% 甲醛或 6.25 µL 16% Formaldehyde, Methanol-Free #12606 和 5 µL Protease Inhibitor Cocktail (200X) #7012 到 1 mL Phosphate Buffered Saline (PBS-1X) pH7.2 (Sterile) #9872 中。使用在制造商标示的有效期内的新鲜甲醛。
- 为每种组织类型制备 1 mL Phosphate Buffered Saline (PBS-1X) pH7.2 (Sterile) #9872 + 5 µL Protease Inhibitor Cocktail (200X) #7012,并置于冰上。
- 每 1 mL 固定缓冲液制备 100 µL Glycine Solution (10X) #7005。
- 为每次抗体反应称量 1 mg 新鲜组织,并为输入样本额外称量 1 mg 组织。确保包含阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 和阴性对照 Rabbit (DA1E) Monoclonal Antibody IgG Isotype Control (CUT&RUN) #66362 的反应。
- 将组织样本放入盘中,并使用干净的手术刀或剃须刀片切碎。将培养皿置于冰上保存。重要的是将组织置于低温下以防止蛋白质降解。
- 将切碎的组织立即转移到 1 mL 固定缓冲液中,并旋转试管以混合。
- 室温孵育 2 分钟。
- 每 1 mL 固定缓冲液添加 100 µL Glycine Solution (10X) #7005 以停止交联。旋转试管混匀并置于室温下孵育 5 分钟。
- 在 4°C 下将组织以 2,000 x g 的离心力离心 5 分钟,然后去除并丢弃液体。
- 用 1 mL 1X PBS + PIC 重悬组织。
- 在 4°C 下以 2,000 x g 的离心力离心 5 分钟,去除并丢弃液体,然后进行步骤 9。(安全停止)或者,固定的组织沉淀物可以在解离前于 -80°C 下储存长达 6 个月。
- 用 1 mL 1X 洗涤缓冲液(+ 亚精胺 + PIC)重悬组织,并将样品转移到 Dounce 匀浆器中。
- 用 20-25 冲程将组织块分解成单细胞悬液,直到观察不到组织块。
- 将细胞悬液转移到 1.5 mL 试管中,并在室温下以 3,000 x g 的离心力离心 3 分钟,从细胞中去除并丢弃上清液。
- 用 1 mL 1X 洗涤缓冲液(+ 亚精胺 + PIC)重悬细胞沉淀物。
- 在室温下将细胞悬液以 3,000 x g 的离心力离心 3 分钟,然后去除并丢弃液体。
- 重复步骤 12 和 13 一次,再次洗涤细胞沉淀物。
- 对于每次反应,添加 100 µL 1X 洗涤缓冲液(+ 亚精胺 + PIC),并轻轻上下吸打来重悬细胞沉淀物。
- 转移 100 µL 细胞到一个新试管中,置于 4°C 下储存,直至进行到第七部分。这是输入样品。
- 立即进入第三部分。
注:对于某些转录因子或辅因子,或对于诸如纤维组织之类难以处理的组织类型,每反应最多可使用 5 mg 组织而无需按比例增加试剂用量。
注:我们建议对组织进行轻微固定,因为这种条件最适合大多数组织类型和蛋白质靶标。但是,如果需要新鲜组织,请跳过步骤 3 到 8,直接进行步骤 9。
注:此体积的固定缓冲液足以处理多达 50 mg 的组织。如果处理的量 >50 mg,则在步骤 7 中相应地增加固定缓冲液和 1X PBS + PIC 溶液的用量。
注:对于难以处理的组织类型(如纤维组织)或低丰度和/或弱结合转录因子或辅因子,将甲醛固定延长至 10 分钟可能会改善结果。
注:在随后的实验步骤中,将在 55°C 下孵育输入样品,因此建议使用一根可封盖的 1.5 mL 试管,以减少孵育过程中的蒸发。
三、添加 Drosophila Spike-In Nuclei Control 用于样品归一化
开始之前:
在室温下解冻 Drosophila Spike-In Nuclei Control。每瓶均提供足够用于最多 8 次反应的 spike-in 核,基于推荐体积:每含有 100,000 至 250,000 个细胞或 1 至 2.5 mg 组织的反应加入 20 µL spike-in 核。
若一个实验中需要用到多瓶 Drosophila Spike-In Nuclei Control,则将解冻的各瓶合并在一起,以确保每反应添加的 spike-in 核数量一致。
尽量减少 Drosophila Spike-In Nuclei Control 的冷冻和解冻次数。分装成小体积份数,以将冻融循环限制在 4 次或更少。超出此限度可能会降低果蝇基因组的比对率,并且需要增加每反应中的 spike-in 核用量;例如,经过 9 次冻融循环后,果蝇比对率可能降低约 50%。
- 对于每次反应,将第二部分 A 中的步骤 6、第二部分 B 中的步骤 8或第二部分 C 中的步骤 15 中制备的 100 µL 细胞悬液分装到单独的 1.5 mL 试管中。
- 向每个含有 100,000 至 250,000 个细胞或 1-2.5 mg 组织的反应中加入 20 µL Drosophila Spike-In Nuclei Control。此 spike-in 核与细胞的比率通常可使果蝇 spike-in 归一化读数占总测序读数的约 0.5%-10%。
- 对于输入量低于 100,000 个细胞或 1 mg 组织的情况,按比例减少 Drosophila Spike-In Nuclei Control 的体积,维持每 100,000 个细胞或每 1 mg 组织对应 20 µL 的比率(例如,对于 50,000 个细胞,使用 10 µL 的体积,对于 5,000 个细胞,使用 1 µL 的体积)。
- 此外,如果靶标丰度较低(例如某些转录因子或辅因子),则 spike-in 核的用量可能需要减少 2 至 10 倍。例如,对于含有 100,000 至 250,000 个细胞或 1 至 2.5 mg 组织的每反应,使用 2 至 10 µL Drosophila Spike-In Nuclei Control。
- 含有 Drosophila Spike-In Nuclei Control的 CUT&RUN 反应可在下游 qPCR 和下一代测序分析 (NGS) 中进行归一化处理。由于这两种检测均使用相同量的 spike-in 核,因此无需重复反应。
- 轻轻地上下吸打混合。
- 立即进行第四部分。
注:Drosophila Spike-In Nuclei Control 的最佳用量因 CUT&RUN 而异,应由用户自行优化,使 spike-in 归一化读数占总测序读数的约 0.5%-10%。
四、Concanavalin A 珠子和一抗的结合
开始之前:
注:推荐用于细胞透化的毛地黄皂苷的量过多,应该足以用于大多数细胞系和组织类型的透化。然而,并非所有细胞系和组织对毛地黄皂苷都表现出相同的敏感性。如果您的特定细胞系或组织在推荐的毛地黄皂苷浓度下不起作用,您可以按照附录 A 中提供的实验步骤来优化条件。毛地黄皂苷处理应能使 >90% 的细胞群发生通透。
注:对于第四至第六部分中的所有孵育步骤,无需通过摇动或旋转来混合样品。只需将试管放在架子上,置于指定温度下即可。混合不会改善检测性能,反而可能导致珠子聚集,或因其粘附在管壁和管盖上而造成珠子损失。
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
将 Digitonin Solution #16359 在 90-100°C 下加热 5 分钟,并确保其完全解冻并呈溶液状态。立即将已融化的 Digitonin Solution #16359 放在冰上。
加热 Protease Inhibitor Cocktail (200X) #7012 和 100X Spermidine #27287。确保两种物质完全融化。请注意,由于含有 DMSO,Protease Inhibitor Cocktail (200X) #7012 置于冰上时会重新冻结。
对于每次反应,制备 1 µL 100X Spermidine #27287 + 0.5 µL Protease Inhibitor Cocktail (200X) #7012 + 2.5 µL Digitonin Solution #16359 + 96 µL Antibody Binding Buffer (CUT&RUN, CUT&Tag) #15338,并放在冰上(每反应 100 µL)。
注:Digitonin Solution #16359 应于 -20°C 下储存。使用过程中请始终置于冰上,并在当天用毕后置于 -20°C 下储存。
- 将第一部分步骤 7 中制备的活化的 Concanavalin A 珠子平衡至室温,并通过轻轻地上下吸打来彻底混合。
- 将 10 µL 珠子悬液添加到第三部分步骤 3 中制备的每管细胞 + Drosophila Spike-In Nuclei Control 悬液中。
- 通过上下吸打将样品充分混合。室温孵育 5 分钟。
- 将试管放在磁力架上,直到溶液变澄清(30 秒至 2 分钟),然后去除并丢弃液体。
- 从磁力架上取下试管。向每个试管中加入 100 µL Antibody Binding Buffer(+ 精胺 + PIC + 毛地黄皂苷),轻轻地上下吸打混合,并置于冰上。
- 向每个反应管中加入适量的检测抗体和 1 µL H2Av Rabbit Monoclonal Antibody,轻轻地上下吸打混合。
- 将样品在 4°C 下孵育 2 小时或在室温下孵育 1 小时。该步骤可以延伸至在 4°C 下过夜。
注:CUT&RUN 需要的检测抗体量会有所不同,应由用户确定。对于阳性对照 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751,向样品中添加 2 µL 抗体。对于阴性对照 Rabbit (DA1E) Monoclonal Antibody IgG Isotype Control (CUT&RUN) #66362,向样品中添加 5 µL 抗体。我们强烈建议使用阴性对照抗体,而不是非抗体对照,因为后者会导致高水平的非特异性 MNase 消化和高背景信号。我们建议在进行 qPCR 和 NGS 分析时,尽可能使用输入样品作为对照。
五. pAG-MNase 酶结合
开始之前:
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
将 Digitonin Solution #16359 在 90-100°C 下加热 5 分钟,并确保其完全解冻并呈溶液状态。立即将已融化的 Digitonin Solution #16359 放在冰上。
加热 Protease Inhibitor Cocktail (200X) #7012 和 100X Spermidine #27287。确保两种物质完全融化。请注意,由于含有 DMSO,Protease Inhibitor Cocktail (200X) #7012 置于冰上时会重新冻结。
对于每次反应,制备 3.2 mL 的毛地黄皂苷缓冲液 (320 µL 10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 + 32 µL 100X Spermidine #27287 + 16 µL Protease Inhibitor Cocktail (200X) #7012 + 80 µL Digitonin Solution #16359 + 2.752 mL Nuclease-free Water #12931)。
对于每次反应,向一个新试管中添加 50 µL 毛地黄皂苷缓冲液(如上所述)和 1.5 µL pAG-MNase 酶来制备 pAG-MNase 预混合物。例如,对于 10 次反应,转移 500 µL 毛地黄皂苷缓冲液到一个新试管中,并添加 15 µL pAG-MNase 酶。轻轻上下吸打以混合,并放在冰上。
注:Digitonin Solution #16359 应于 -20°C 下储存。使用过程中请始终置于冰上,并在当天用毕后置于 -20°C 下储存。
注:此处制备的毛地黄皂苷缓冲液将用于第五和第六部分。
- 将第四部分步骤 7 中的试管放在磁力架上,直到溶液变澄清(30 秒至 2 分钟),然后去除并丢弃液体。
- 从磁力架上取下试管,添加 1 mL 毛地黄皂苷缓冲液(+ 亚精胺 + PIC + 毛地黄皂苷)。通过轻轻上下吸打重悬珠子,确保收集粘在管壁上的所有珠子。
- 将试管放在磁力架上,直到溶液变澄清(30 秒至 2 分钟),然后去除并丢弃液体。
- 从磁力架上取下试管。向每个试管添加 50 µL pAG-MNase 预混合物,并轻轻上下吸打来混合样品。
- 将试管在 4°C 下孵育 1 小时。
- 立即进入第六部分。
六、DNA 消化和释放
开始之前:
! 所有缓冲液的体积都应根据正在进行的 CUT&RUN 反应次数按比例增加。
将 Digitonin Solution #16359 在 90-100°C 下加热 5 分钟,并确保其完全解冻并呈溶液状态。立即将已融化的 Digitonin Solution #16359 放在冰上。
如果从第二部分中的固定材料开始,请确保 10% SDS Solution #20533 完全呈溶液状态。在 37°C 下加热将有助于溶解 SDS 沉淀物。
对于每次反应,制备 150 µL 消化缓冲液(第五部分中制备的 148.5 µL 毛地黄皂苷缓冲液 + 1.5 µL 氯化钙)。使用前置于冰上。
对于每次反应,制备 150 µL 1X Stop Buffer (37.5 µL CUT&RUN 4X Stop Buffer #48105 + 3.75 µL Digitonin Solution #16359 + 0.75 µL RNAse A (10 mg/mL) #7013 + 108 µL Nuclease-free Water #12931)。
注:Digitonin Solution #16359 应于 -20°C 下储存。使用过程中请始终置于冰上,并在当天用毕后置于 -20°C 下储存。
- 将第五部分步骤 5 中的试管放在磁力架上,直到溶液变澄清(30 秒至 2 分钟),然后去除并丢弃液体。
- 从磁力架上取下试管。添加在第五部分中制备的 1 mL 毛地黄皂苷缓冲液(+ 亚精胺 + PIC + 毛地黄皂苷),并通过轻轻地上下吸打来重悬珠子。
- 将试管放在磁力架上,直到溶液变澄清(30 秒至 2 分钟),然后去除并丢弃液体。
- 重复步骤 2 和 3 一次。
- 从磁力架上取下试管。向每个试管中添加 150 µL 消化缓冲液来激活 pAG-MNase,并上下吸打混合。
- 在 4°C 下孵育样品 30 分钟。
- 向每份样品中添加 150 µL 1X Stop Buffer(+ 毛地黄皂苷 + RNAse A),并上下吸打来混合。
- 在 37°C 下孵育试管 10 分钟,不要摇晃,使 DNA 片段进入溶液。
- 以 16,000 x g 的离心力在 4°C 下离心 2 分钟,然后将试管放在磁力架上,直到溶液变澄清(30 秒至 2 分钟)。
- 将上清液转移到新的 2 mL 微量离心管中。这些是富含染色质的样品。
- 要逆转固定细胞或组织样品中的交联,让样品升温至室温并添加 3 µL 10% SDS Solution #20533(0.1% 最终浓度)和 2 µL Proteinase K (20 mg/mL) #10012 到每份样品。
- 涡旋搅拌每个样品并在 65°C 下孵育至少 2 个小时。这次孵育可以延长过夜。孵育后,以 10,000 x g 的离心力快速旋转样品 1 秒,以收集管盖上因蒸发产生的冷凝液。
- 将样品平衡至室温并进行第八部分。(安全停止)或者,样品可于 -20°C 下储存长达 1 周。但是,在 DNA 纯化(第八部分)之前,请确保将样品加热到室温。
注:应在 4°C 的控温板上或冰箱中进行消化。冰的温度可低至 0°C,这可能会限制消化并降低信号。
注:如果使用活细胞或新鲜组织(未固定)进行 CUT&RUN 检测,则跳过步骤 11 和 12,直接进行步骤 13。
注:在随后的实验步骤中,将在 65°C 下孵育固定样品,因此建议使用一根可封盖的 2 mL 试管,以减少孵育过程中的蒸发。
注:如果样品没有预热到室温,SDS 可能会从溶液中沉淀出来。
七、输入样品制备
输入 DNA 的片段化是与下游 NGS 兼容所必需的,但对于下游 qPCR 分析则不是必需的。如果没有超声波仪,我们建议使用未片段化的输入 DNA 进行 qPCR 分析;然而,输入 DNA 应使用苯酚/氯仿提取和乙醇沉淀进行纯化,因为未片段化输入 DNA 的尺寸太大而无法使用 DNA 离心柱进行纯化。如果没有超声波仪并且需要进行下游 NGS 分析,则可以使用 CUT&RUN 正常 IgG 抗体样品作为阴性对照,尽管这并不理想,因为富含 IgG 的正常样品可能会显示非特异性 DNA 富集。或者,可以在 cst-science.com/CUT-RUN-input-digestion 上获取使用 MNase 的输入 DNA 片段化实验步骤。
开始之前:
! 所有缓冲液的体积都应根据正在制备的输入样品的数量按比例增加。
加热 DNA Extraction Buffer #42015。确保完全澄清。
对于每份输入样品,制备 2 µL Proteinase K (20 mg/mL) #10012 + 0.5 µL RNAse A (10 mg/mL) #7013 + 197.5 µL CUT&RUN DNA Extraction Buffer #42015(每份输入样品共 200 µL)。
- 将 200 µL DNA Extraction Buffer (+ Proteinase K + RNAse A) 添加到 100 µL 输入样品中,这些样品来自第二部分 A 中的步骤 7、第二部分 B 中的步骤 9 或第二部分 C 中的步骤 16。上下吸打混合。
- 在 55°C 下孵育试管 1 小时,以高达 1,200 rpm 的速度进行中度至剧烈振荡。
- 将试管放在冰上 5 分钟,让样品完全冷却。
- 对输入样品进行超声处理,以裂解细胞并破碎染色质。在两次脉冲之间将样品放在冰上孵育 30 秒。
- 在 4°C 下以 18,500 x g 的离心力离心 10 分钟,以澄清裂解物。将上清液转移至一个新的 2 mL 微量离心管中。
- 立即进行第八部分“DNA 纯化”。(安全停止)或者,输入样品可于 -20°C 下储存长达 1 周。但是,在 DNA 纯化程序(第八部分)之前,请确保将输入样品加热到室温。
注:可能需要按照附录 B 中的实验步骤测试不同的超声波仪功率设置和/或持续时间,以根据经验确定超声处理条件。最佳超声处理条件将产生大小为 100-600 bp 的染色质片段。使用配备 1/8 英寸探头的 VirTis Virsonic 100 超声波均质器/超声波仪,在设置 6 档位下,进行五次持续 15 秒脉冲的超声处理,可充分碎裂输入染色质。
八、DNA 纯化
如第八部分 A 中所述,使用 DNA 离心柱可从输入和富集染色质样品中纯化 DNA,或如第八部分 B 中所述,使用苯酚/氯仿提取后再行乙醇沉淀方法进行纯化。使用 DNA 离心柱纯化简单快速,可很好地回收 35 bp 以上的 DNA 片段(图 7A,泳道 2)。苯酚/氯仿提取之后再行乙醇沉淀会更加困难,但能很好地回收 35 bp 以下的 DNA 片段(图 7A,泳道 3);但如图 7B 所示,大多数在 CUT&RUN 检测中产生的 DNA 片段大于 35 bp。因此,DNA 离心柱提供一种快速简单的方法来纯化 > 98% 的所有 CUT&RUN DNA 片段。
在 NGS 分析之前,使用基于 picogreen 的 DNA 定量测定法可以对纯化的 DNA 进行定量。对于含 100,000 个细胞的 CUT&RUN 反应,预期 DNA 产量范围为:针对转录因子和辅因子,每反应 0.5-10 ng;针对组蛋白修饰,每反应 1-20 ng。
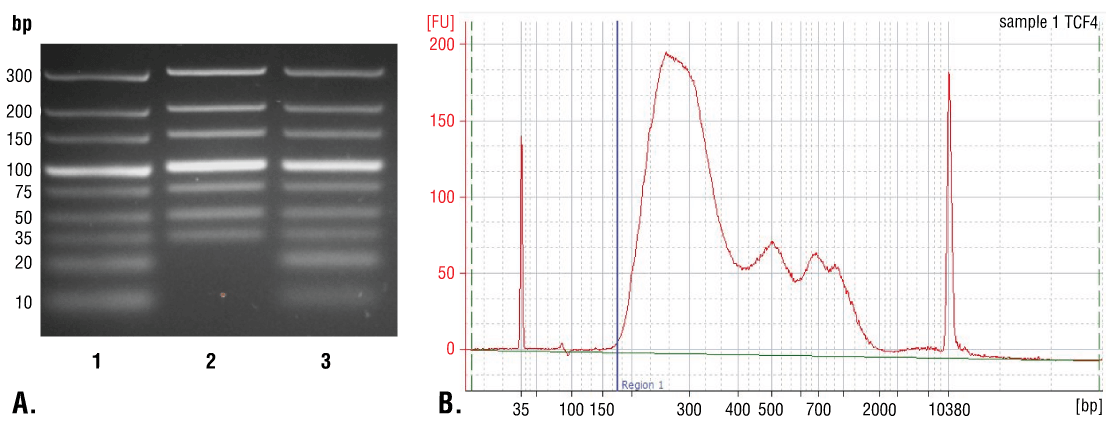
图 7. 比较使用离心柱或苯酚/氯仿提取后再行乙醇沉淀方法进行的 DNA 纯化。(A) 对低量程 DNA 标准品混合物(泳道 1,未纯化)使用 DNA Purification Buffers and Spin Columns (ChIP, CUT&RUN, CUT&Tag) #14209(泳道 2)或苯酚/氯仿提取继而乙醇沉淀法(泳道 3)纯化,并通过在 4% 琼脂糖凝胶上电泳来分离。如图所示,苯酚/氯仿提取后再行乙醇沉淀能有效回收所有大小的 DNA 片段,而 DNA 离心柱仅能回收 ≥ 35 bp 的 DNA 片段。(B) 在使用 TCF4/TCF7L2 (C48H11) Rabbit Monoclonal Antibody #2569 的 CUT&RUN 检测中使用苯酚/氯仿提取继而乙醇沉淀法来纯化 DNA。使用一台 Bioanalyzer (Agilent Technologies) 分析文库中 DNA 片段的大小。构建期间向文库添加的接头和条码序列占了 140 bp 片段长度。因此,起始的 35 bp DNA 片段长度将在文库制备后变为 175 bp(用蓝色垂直线标注)。如图所示,CUT&RUN 富集的总 DNA 片段中不到 2% 短于 175 bp(起始长度 35 bp),这提示 DNA 纯化离心柱足以捕获 > 98% 的总 CUT&RUN DNA 片段。
A. 使用离心柱进行 DNA 纯化
注:使用 DNA Purification Buffers and Spin Columns (ChIP, CUT&RUN, CUT&Tag) #14209(未在本试剂盒中包含)以及下面改良的实验步骤,可以从输入和富集的染色质样品中纯化 DNA。步骤 1-5 已修改,以反映向 300 µL 输入和富集染色质样品中添加 5 倍体积 (1.5 mL) DNA 结合缓冲液的要求。
开始之前:
!! 使用前添加 24 mL 乙醇 (96-100%) 到 DNA 洗涤缓冲液中。在第一组 DNA 纯化之前,只需要添加一次。
针对每份需纯化的富集染色质样品或输入样品,制备一个 DNA 纯化和收集管。
- 向每份输入或富集染色质样品中添加 1.5 mL DNA 结合缓冲液,并上下吸打来混合。
- 从在步骤 1 中制备的每份样品中取 600 µL 并将其转移至收集管中的 DNA 离心柱内。
- 用微量离心机以 18,500 x g 的离心力离心 30 秒。
- 从收集管取出离心柱并丢弃液体。将离心柱放在空的收集管中。
- 重复步骤 2-4,直到步骤 1 中的整个样品已经过离心柱旋转。将离心柱放回空的收集管。
- 向收集管中的离心柱添加 750 µL DNA 洗涤缓冲液。
- 用微量离心机以 18,500 x g 的离心力离心 30 秒。
- 从收集管取出离心柱并丢弃液体。将离心柱放在空的收集管中。
- 用微量离心机以 18,500 x g 的离心力离心 30 秒。
- 丢弃收集管和液体。保留离心柱。
- 向每个离心柱添加 50 µL DNA 洗脱缓冲液,并放在干净的 1.5 mL 管中。
- 用微量离心机以 18,500 x g 的离心力离心 30 秒,以洗脱 DNA。
- 取出并丢弃 DNA 离心柱。洗脱物即纯化的 DNA。(安全停止)样品可于 -20°C 下储存长达 6 个月。
注:每 1 体积的样品应使用 5 体积的 DNA Binding Buffer。
B. 使用苯酚/氯仿提取和乙醇沉淀方法进行 DNA 纯化
注:以下试剂是苯酚/氯仿提取和乙醇沉淀所必需的,不包含在本试剂盒中:酚/氯仿/异戊醇 (25:24:1)、氯仿/异戊醇 (24:1)、3M 乙酸钠 (pH 5.2)、20 mg/mL 糖原、100% 乙醇、70% 乙醇和 1X TE 缓冲液或 Nuclease-free Water #12931。
- 向每份输入和富集染色质样品中添加 300 µL 苯酚/氯仿/异戊醇 (25:24:1),并涡旋搅拌 30 秒充分混合。
- 使用微量离心机以 16,000 x g 的离心力离心 5 分钟来分层。小心地将大部分顶部水层(避开中间层)转移到一个新管中。
- 向水相样品中添加 300 µL 氯仿/异戊醇 (24:1),并涡旋搅拌 30 秒充分混合。
- 使用微量离心机以 16,000 x g 的离心力离心 5 分钟来分层。小心地将大部分顶部水层(避开中间层)转移到一个新管中。
- 向每份水相样品中添加 25 µL 3M 乙酸钠 (pH 5.2)、1 µL 20 mg/mL 糖原和 300 µL 100% 乙醇,并涡旋搅拌 30 秒混合。
- 将样品在 -80°C 下孵育 1 小时或在 -20°C 下孵育过夜来使 DNA 沉淀。
- 用微量离心机在 4°C 下以 16,000 x g 的离心力离心 5 分钟来使 DNA 沉淀。
- 小心地去除并丢弃上清液,用 70% 乙醇洗涤沉淀物。
- 用微量离心机在 4°C 下以 16,000 x g 的离心力离心 5 分钟来使 DNA 沉淀。
- 倒出上清液,并风干沉淀物。
- 在 50 µL 1X TE 缓冲液或 Nuclease-free Water #12931 中重悬沉淀物。这是纯化的 DNA。(安全停止)样品可于 -20°C 下储存长达 6 个月。
九、通过 qPCR 进行 DNA 定量
建议:
本试剂盒中包含的果蝇归一化引物组对黑腹果蝇 CG3402 基因具有特异性,可用于定量来自 Drosophila Spike-In Nuclei Control 的信号以进行样品归一化。
本试剂盒中包含的额外对照引物对人或小鼠 RPL30 基因(#7014 或 #7015)具有特异性,可用来对 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 样品进行定量实时 PCR 分析。如果对其他物种进行 CUT&RUN,则用户需要为该物种设计相应的对照引物,并确定最佳 PCR 条件。
PCR 引物选择至关重要。对于 CUT&RUN,PCR 扩增子大小应大约为 60-80 bp 长。引物的最佳熔解温度应设计为 60°C 左右,GC 含量应在 50% 左右。
2 µL 纯化的 DNA 足以对组蛋白、转录因子和辅因子的靶基因进行 qPCR 介导的定量。
建议使用热启动 Taq 聚合酶以最大限度降低非特异性 PCR 产物的风险。
使用过滤式吸液头来尽量减少污染风险。
- 标记适当数量、兼容待用 PCR 仪型号的 PCR 管或 PCR 平板。PCR 反应应当包括阳性对照三甲基组蛋白 H3 Lys4 样品、阴性对照兔 IgG 样品、无 DNA 的管(作为 DNA 污染的对照)以及 DNA 输入样品。如果需要,可以使用输入 DNA 的梯度稀释物(未稀释 - 100% 输入,1:5 - 20% 输入,1:25 - 4% 输入,1:125 - 0.8% 输入)创建标准曲线,以确定扩增效率及定量每个免疫富集的样品中 DNA 的量。
- 向每个 PCR 管或 PCR 板的孔中添加 2 µL 适宜 DNA 样品。
- 按下文所述的步骤制备主反应混合物。每次 PCR 反应设置 2-3 个重复样品。添加足量试剂来弥补损耗的体积(1-2 次额外反应)。向每个 PCR 反应管或各孔中添加 18 µL 反应混合物。
- 启动以下 PCR 反应程序:
- 使用实时 PCR 仪的自带软件,分析定量 PCR 结果。或者,可以使用输入样品百分比方法和以下所示的公式,手动计算 IP 效率。采用这种方法,从每个抗体反应获得的信号表述为占总输入染色质的百分比。如果使用 DNA 输入样品的梯度稀释物,则绘制并利用标准曲线比照输入百分比(100%、20%、4%、0.8%)的 Log(10) 计算从每个抗体反应所获得的信号。
- 输入百分比 = 100% x 2(C[T] 100% 输入样品 – C[T] IP 样品)
- C[T] = Ct = Cq = PCR 反应的平均循环阈值
- 对于样品归一化,从果蝇归一化引物组中选择 C[T] 值最低的样品作为参考样品(例如,下表示例中的样品 1)。使用提供的方程式计算所有其他样品的归一化系数。使用计算所得的归一化系数调整检测引物组的信号。
注:仅应使用果蝇归一化引物集分析 CUT&RUN 样品,因为输入 DNA 不含用作 qPCR 模板的 Drosophila Spike-In Nuclei Control。
|
试剂 |
1 次 PCR 反应所需的体积 (18 µL) |
|---|---|
| Nuclease-free Water #12931 | 6 µL |
| 5 µM 引物 | 2 µL |
| SimpleChIP® Universal qPCR Master Mix #88989 | 10 µL |
| a. | 初始变性 | 95°C,3 分钟 |
| b. | 变性 | 95°C,15 秒 |
| c. | 复性和延伸 | 60°C,60 秒 |
| d. | 重复步骤 b 和 c,共循环 40 次。 |
使用 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751 和 SimpleChIP® Human β-Actin Promoter Primers #13653 进行 qPCR 检测的样品归一化示例(参见图 8)
| Dm 归一化引物组的 C[T]值 | **qPCR 的标准化系数 | 归一化前的信号(% 输入) | 归一化后的信号(% 输入) | |
| 样品 1 | 26.72 | 2(26.72-26.72)=1.00 | 0.99% | 0.99%/1.00=0.99% |
| 样品 2 | 27.61 | 2(26.72-27.61)=0.54 | 0.61% | 0.61%/0.54=1.13% |
| 样品 3 | 29.29 | 2(26.72-29.29)=0.17 | 0.20% | 0.20%/0.17=1.18% |
| 样品 4 | 31.08 | 2(26.72-31.08)=0.05 | 0.06% | 0.06%/0.05=1.20% |
**qPCR 的归一化系数 = 2(C[T] 参考样品 – C[T] 其他样品)
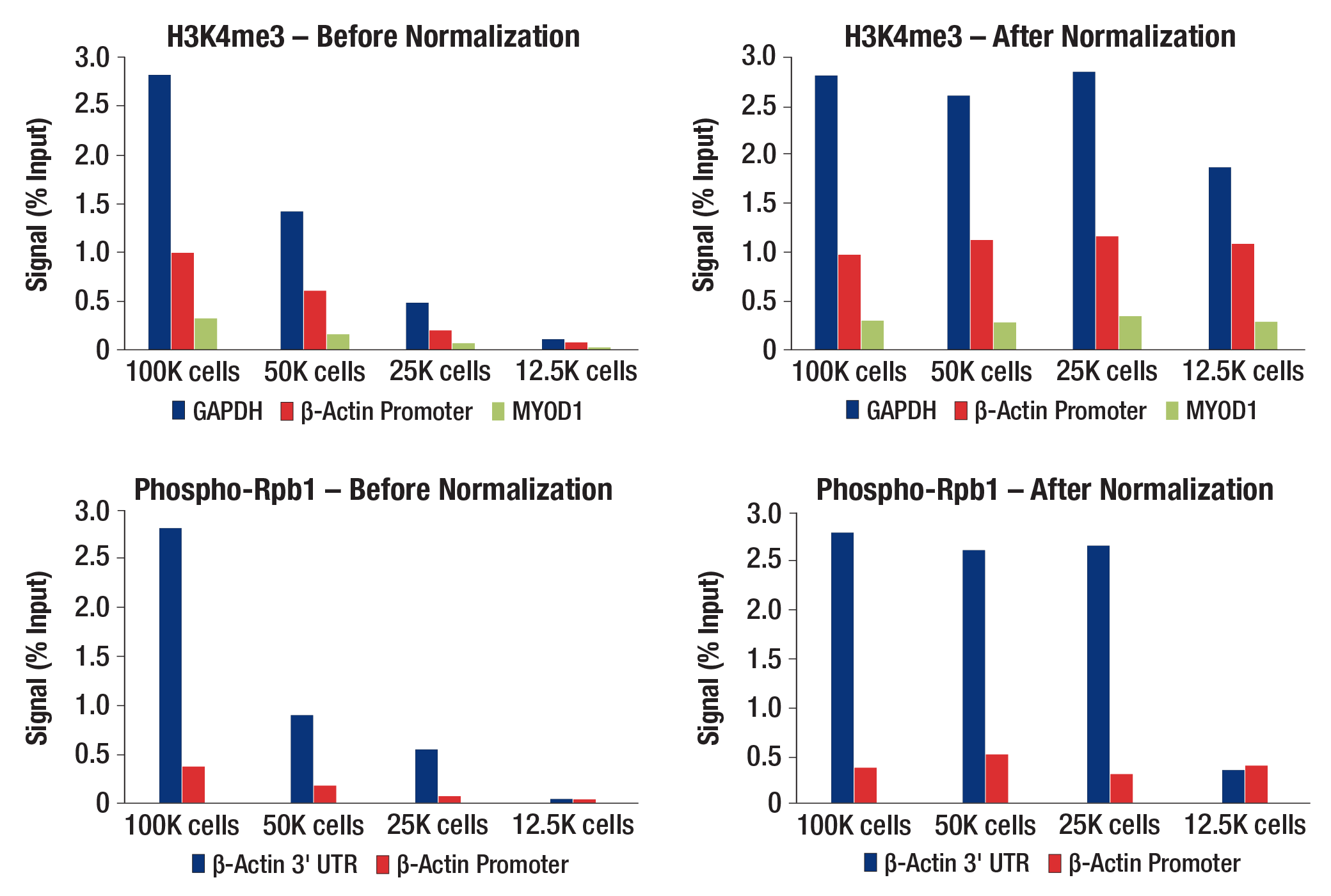
图 8. 利用 Drosophila Spike-In Nuclei Control 对 CUT&RUN 信号进行归一化处理,以用于 qPCR 分析。使用数量递减的 HCT 116 细胞与 Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit Monoclonal Antibody #9751(顶部小图)或 Phospho-Rpb1 CTD (Ser2) (E1Z3G) Rabbit Monoclonal Antibody #13499(底部小图)进行 CUT&RUN 测定。使用 human GAPDH exon 1 primers、SimpleChIP® Human beta-Actin Promoter Primers #13653、SimpleChIP® Human beta-Actin 3' UTR Primers #13669 和 SimpleChIP® Human MyoD1 Exon 1 Primers #4490 进行实时 PCR 来对富集的 DNA 进行定量。每份样品中的免疫沉淀 DNA 数量表示为与输入染色质总量(100,000 个细胞的输入百分比)相对应的信号。左图中为非标准化的富集结果。在每次反应中,按照与起始细胞数量成比例的方式添加 Drosophila Spike-In Nuclei Control。根据各样品中使用果蝇归一化引物组从 Drosophila Spike-In Nuclei Control 中获得的 qPCR 信号差异,将 CUT&RUN 信号归一化至含有 100,000 个细胞的样品。右图显示了标准化后的富集。值得注意的是,当 CUT&RUN 检测因输入量极低而失败时,归一化后的信号能准确地反映失败这一结果,而不会产生人为偏高的信号,这充分证明了该归一化策略的可靠性和稳健性(参见使用 12.5K 个细胞的 Rpb1 数据)。
十. NGS 文库构建
用本试剂盒制备的免疫富集 DNA 样品直接兼容 NGS。要构建下游 NGS DNA 文库,请使用与您的下游测序平台相容的 DNA 文库制备实验步骤或试剂盒。对于在 Illumina Systems 平台上测序,我们建议按照 CUT&RUN DNA 的实验步骤,使用 DNA Library Prep Kit for Illumina Systems (ChIP-seq, CUT&RUN) #56795 和 Multiplex Oligos for Illumina Systems (ChIP-seq, CUT&RUN) #29580 或 #47538 进行测序。
由于 CUT&RUN 中产生的背景信号很低,因此每份样品 500 万个读数的测序深度对于组蛋白修饰和转录因子而言通常已经足够了。如果每份样品的测序深度大于 1,500 万,则读数的重复率显著增加。如果每份样品的测序深度低于 200 万,则信噪比会降低。
对于少于 20,000 个起始细胞数,在 NGS 读数中获得较低的比对率或较高的重复率是很常见的。如果发生这种情况,我们建议增加测序深度以获得足够数量的唯一比对读数,用于下游数据分析。
对于样品归一化,将所有样品的 CUT&RUN 测序数据同时比对至检测基因组(例如人、小鼠或其他物种)和黑腹果蝇基因组 计算每份样品中唯一的 spike-in 读数与总唯一比对读数的比率。选择比率最低的样品(如下表中的样品 4)作为参考样品,并使用提供的公式计算其他样品的归一化系数。在生物信息学分析过程中,通过缩放 bigWig 文件(例如使用 deepTools bamCoverage)或对每份样品比对至检测基因组的唯一读数进行降采样处理,来应用这些系数。如果各样品间的测序深度一致,也可以使用 spike-in 读数绝对值而非其与总读数的比率来计算归一化系数。
使用 Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit Monoclonal Antibody #9733 在细胞滴定实验中进行 NGS 检测的样品归一化示例(参见图 2)
| 比对至 Spike-In 基因组 (dm6) 的唯一读数数量 | 比对至检测参考基因组 (hg38) 的唯一读数数量 | Spike-In 唯一读数占唯一比对总读数的比率 | NGS 的标准化系数 | |
| 样品 1 | 262,459 | 1,160,970 | 262,459/(262,459 + 1,160,970) = 0.184 | 0.034/0.184= 0.19 |
| 样品 2 | 113,312 | 1,027,248 | 113,312/(113,312 + 1,027,248) = 0.099 | 0.034/0.099= 0.34 |
| 样品 3 | 184,441 | 2,998,350 | 184,441/(184,441 + 2,998,350) = 0.058 | 0.034/0.058= 0.59 |
| 样品 4 | 66,880 | 1,901,014 | 66,880/(66,880 + 1,901,014) = 0.034 | 0.034/0.034= 1 |
NGS 归一化系数 = 参考样品的 spike-in 唯一读数占唯一比对总读数的比率 / 其他样品的 spike-in 唯一读数占唯一比对总读数的比率
附录 A:测试细胞对毛地黄皂苷的敏感性
在 CUT&RUN 实验步骤中,向缓冲液添加毛地黄皂苷促进了细胞膜透化以及一抗和 pAG-MNase 酶进入细胞和胞核。因此,缓冲液中有足量的毛地黄皂苷对抗体和酶的成功结合以及靶向基因位点的消化至关重要。不同细胞系对毛地黄皂苷透化细胞显示不同的敏感性。虽然本实验步骤中建议的毛地黄皂苷量应足以对大多数细胞系或组织进行透化,但您可以使用本实验步骤测试您的特定细胞系或组织。我们发现,添加过量的毛地黄皂苷对本实验没有损害,因此无需生成浓度曲线。所以,进行快速测试以确定建议的毛地黄皂苷量是否适合你的细胞系就足够了。
开始之前:
将 Digitonin Solution #16359 在 90-100°C 下加热 5 分钟。确保其完全解冻。立即将已融化的 Digitonin Solution #16359 放在冰上。
对于每个细胞或组织样品,制备 100 µL 洗涤缓冲液 (10 µL 10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 + 90 µL Nuclease-free Water #12931)。此检测无需添加 100X Spermidine #27287 或 Protease Inhibitor Cocktail (200X) #7012。
注:Digitonin Solution #16359 应于 -20°C 下储存。使用过程中请始终置于冰上,并在当天用毕后置于 -20°C 下储存。
- 在一个 1.5 mL 管中,收集 10,000 至 100,000 个细胞。对于组织,从 1 mg 组织中收集分解的细胞(第二部分 C 中的步骤 1-15)。
- 在室温下以 600 x g 的离心力离心 3 分钟,然后去除并丢弃液体。
- 用 100 µL 洗涤缓冲液重悬细胞沉淀物。
- 将 2.5 µL Digitonin Solution #16359 添加到每次反应中,并在室温下孵育 10 分钟。
- 将 10 µL 细胞悬液与 10 µL 0.4% 台盼蓝染液混合。
- 使用血细胞计数器或细胞计数器计算染色细胞的数量和细胞总数。充分通透导致 >90% 的细胞着染台盼蓝。
- 如果被台盼蓝染色的细胞不足 90%,则增加毛地黄皂苷缓冲液中添加的 Digitonin Solution #16359 的量,并重复步骤 1-6,直到 >90% 的细胞被通透和染色。在第四至第六部分中采用这一 Digitonin Solution #16359 的量。
注:如果肉眼看不到细胞沉淀物,我们建议在步骤 2 中对细胞悬液进行初始离心后,在不干扰细胞沉淀物的情况下去除尽可能多的细胞培养基,并为每反应保留 ≤ 40 µL 的细胞培养基。然后在步骤 3 中,向细胞悬液中加入足够的 1X 洗涤缓冲液,使总体积达到 100 µL。
附录 B:对输入样品进行超声处理优化
建议对输入 DNA 样品进行超声处理,因为只有经过片段化的基因组 DNA (< 10 kb) 才能使用 DNA 纯化离心柱进行纯化。此外,片段化的基因组 DNA (< 1 kb) 可在 NGS 分析中用作阴性对照。应优化超声处理步骤,以使 DNA 输入的长度为 100-600 bp。
我们建议使用输入样品进行 NGS,因为它能方便且无偏倚地呈现细胞基因组的情况。虽然 IgG 样品也可在 NGS 中用作阴性对照,但由于存在非特异性结合,它可能会显示基因组特定区域出现富集。非片段化input DNA 可用于 qPCR 分析。但必须使用苯酚/氯仿提取后再行乙醇沉淀方法来纯化非片段化的 DNA。
开始之前:
! 所有缓冲液的体积都应根据正在制备的输入样品的数量按比例增加。
将 CUT&RUN DNA Extraction Buffer #42015 置于室温下加热,确保其完全融化并呈溶液状态。
对于每份输入样品,制备 2.1 mL 1X 洗涤缓冲液 (210 µL 10X Wash Buffer (CUT&RUN, CUT&Tag) #31415 + 1.89 mL Nuclease-free Water #12931),并让其平衡到室温,以最大程度地减少细胞应激。此检测无需添加 100X Spermidine #27287 或 Protease Inhibitor Cocktail (200X) #7012。
对于每份输入样品,制备 2 µL Proteinase K (20 mg/mL) #10012 + 0.5 µL RNAse A (10 mg/mL) #7013 至 197.5 µL CUT&RUN DNA Extraction Buffer #42015(每份输入样品 200 µL)。
- 在 1.5 mL 试管中,收集与您在 CUT&RUN 实验中用于输入的相同数量的细胞(5,000 到 100,000 个细胞),用于待测试的每个超声处理条件。对于组织,从用于 CUT&RUN 实验(第二部分 C 中的步骤 1-15)输入的相同数量的组织中收集分离的细胞,用于待测试的每个超声处理条件。
- 在室温下以 600 x g 的离心力离心 3 分钟,然后去除并丢弃液体。
- 用 1 mL 1X 洗涤缓冲液通过轻轻上下吸打来重悬细胞沉淀物。
- 在室温下以 600 x g 的离心力离心 3 分钟,然后去除并丢弃液体。
- 重复步骤 3 和 4 一次,再次洗涤细胞沉淀物。
- 对于每个被测试的超声处理条件,加入 100 µL 的 1X 洗涤缓冲液,并通过轻轻上下吸打重悬细胞沉淀物。
- 对于每个超声处理的条件,分装 100 µL 细胞悬液到一个新试管中。
- 向每份样品添加 200 µL CUT&RUN DNA Extraction Buffer (+ Proteinase K + RNAse A),并通过上下吸打来混合。
- 在 55°C 下孵育试管 1 小时,以高达 1,200 rpm 的速度进行中度至剧烈振荡。
- 将试管放在冰上 5 分钟,让样品完全冷却。
- 通过设置时间梯度实验(采用 15 秒脉冲超声循环,且循环次数逐步增加),为您的超声波破碎仪确定最优超声条件。确保在两次脉冲之间将样品放在冰上孵育 30 秒。
- 使用微量离心机在 4°C 下以 18,500 x g 的离心力离心 10 分钟,以澄清裂解物。将上清液转移至一个新的 2 mL 微量离心管中。
- 按照第八部分的说明,使用 DNA 纯化离心柱或使用苯酚/氯仿提取后再行乙醇沉淀方法来纯化 DNA 样品。
- 从离心柱中洗脱 DNA,或在 30 µL 1X TE 缓冲液或 Nuclease-free Water #12931 中重悬 DNA 沉淀物。
- 通过电泳确定 DNA 片段大小。将 > 15 µL 样品上样到含 100 bp DNA 标准品的 1% 琼脂糖凝胶上。建议使用无染料上样缓冲液(30% 甘油)以便更好地观察凝胶上的 DNA 拖尾。
- 选择能产生最佳 DNA 片段大小 (100-600 bp) 的超声处理条件,并用于第七部分步骤 4 中的输入样品的制备。如果未达到最佳声处理条件,则增加或减少超声波仪的功率设置或超声处理循环次数,并重复超声处理时程实验。
注:如果在处理低细胞数(< 100,000 个细胞)时肉眼看不到离心后的细胞沉淀物,我们建议跳过下面的洗涤步骤 3-5。在步骤 2 中对细胞悬液进行初始离心后,在不干扰细胞沉淀物的情况下去除并丢弃尽可能多的细胞培养基,并为每反应保留 ≤ 40 µL 的细胞培养基。然后在步骤 6 中,将足够的 1X 洗涤缓冲液添加到细胞悬液中,以便在每个被测试的超声处理条件下达到 100 µL 的体积。
注:步骤 9 中将在 55°C 下孵育样品,因此建议使用一根可封盖的 1.5 mL 试管,以减少孵育过程中的蒸发。
附录 C:疑难排解指南
如需详细的故障排除指南,请访问 //cst-science.com/troubleshooting-CUT+-RUN
附录 D:在线资源
有关 CUT&RUN 检测的可视化实验步骤、研讨会、海报、博客、常见问题解答和更多信息,请访问我们的 CUT&RUN 资源中心,网址为 cellsignal.com/applications/cut-and-run,或扫描下面的二维码。
